MetFragWeb 2010
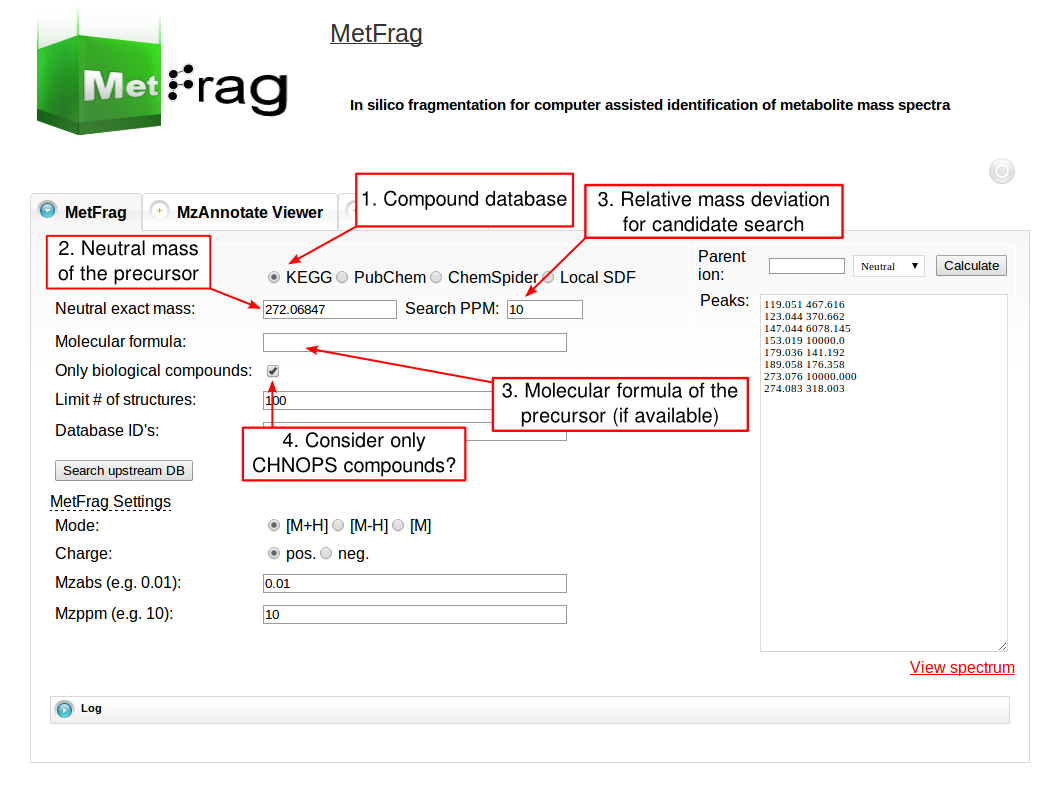
The web tool of MetFrag 2010 has several fields for setting parameters that depend on your experimental conditions during the MS acquisition. For the beta version of the web tool, which includes an in silico derivatised verions of the KEGG database, visit this page. At first the parameters for the structure database query have to be defined under Database Settings. The structure database has to be selected that can be queried either by a monoisotopic mass, a molecular formula or comma separated database dependent identifiers. The first has to be defined together with a mass deviation in ppm.
After setting the parameters for the database query the “Search upstream DB” button can be pushed to submit the database query.
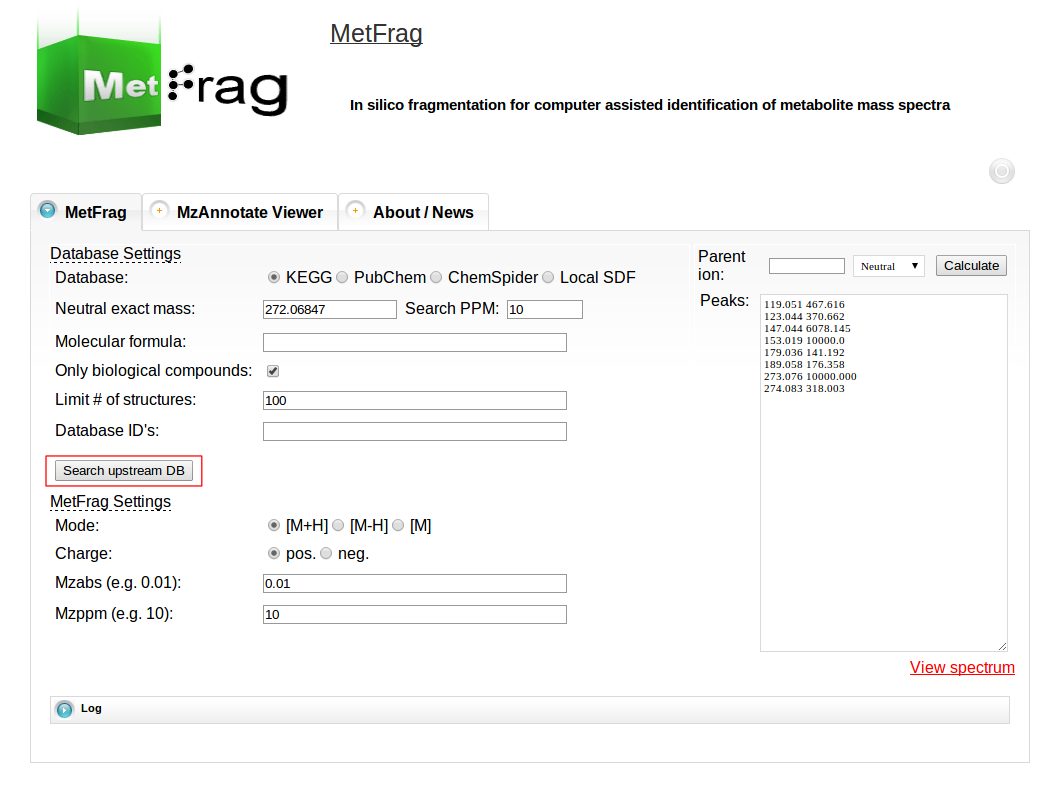
After a short processing time the number of candidates found in the database with the defined filter criteria is displayed.
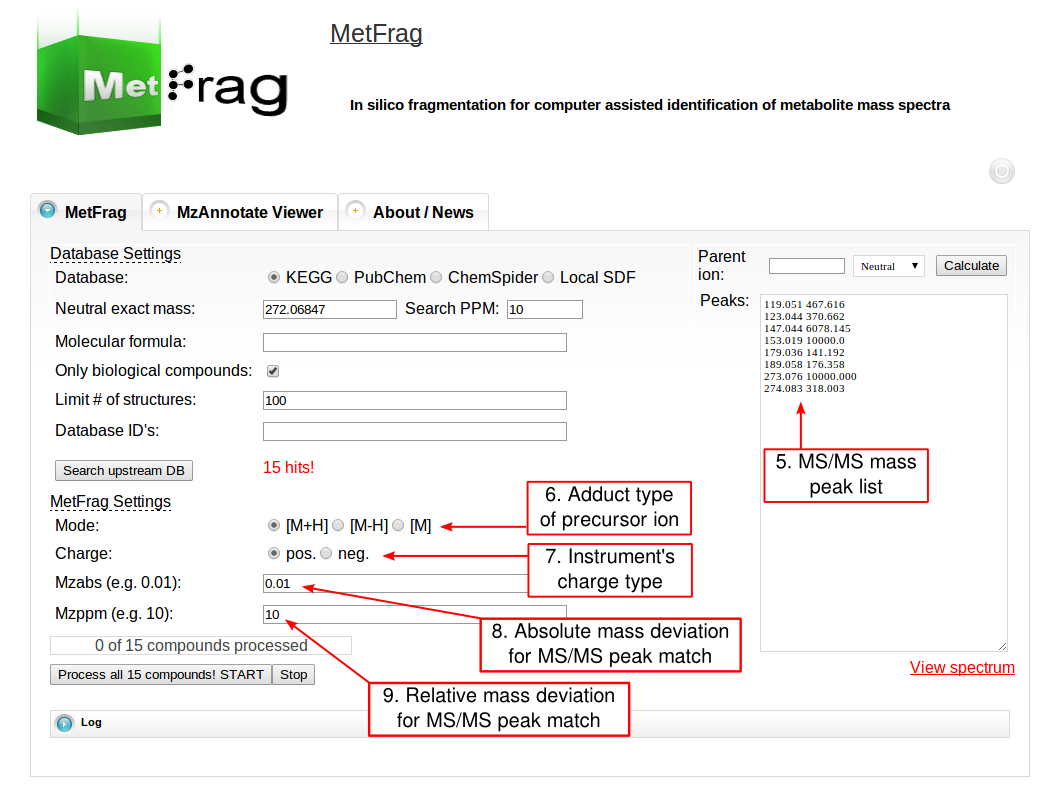
Then the parameters for MetFrag processing have to be defined under the MetFrag Settings. This includes the ionisation mode as together with the charge the data has been acquired. Additionally, the a relative and absolute mass deviation has to be defined with which in silico generated fragments will be mapped to the experimental data defined in the “Peaks” text field. After all parameters are defined correctly, the button “Process all x compounds! START” has to be pushed to start the processing.
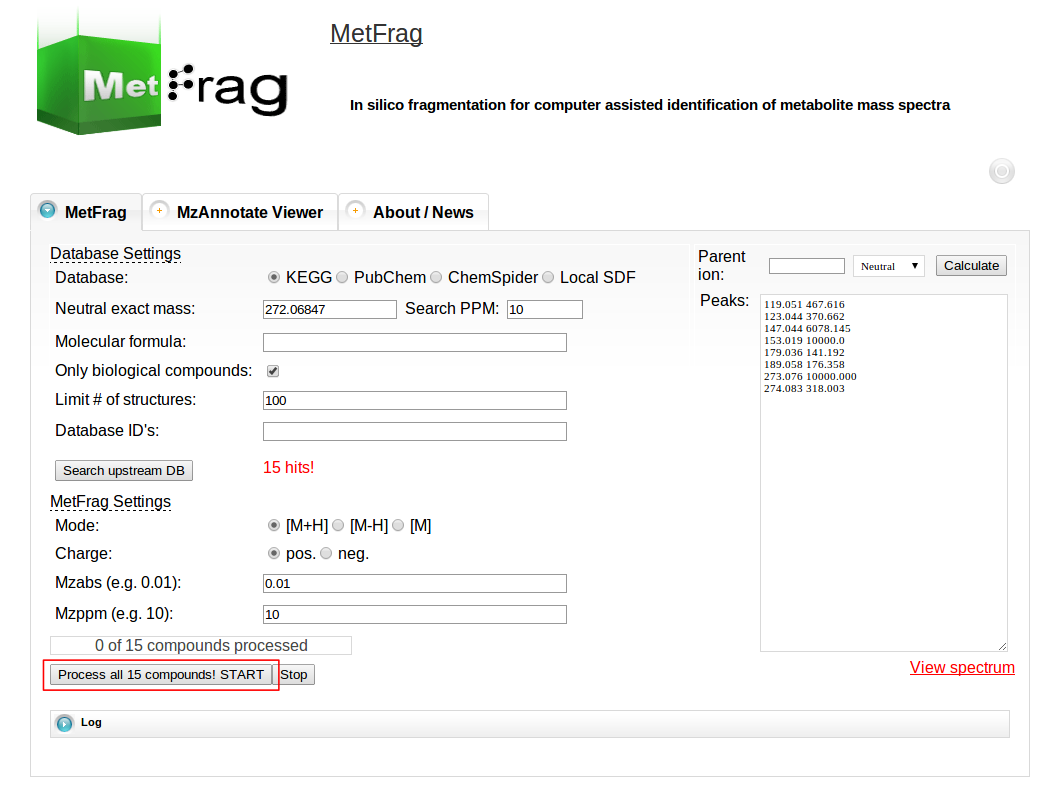
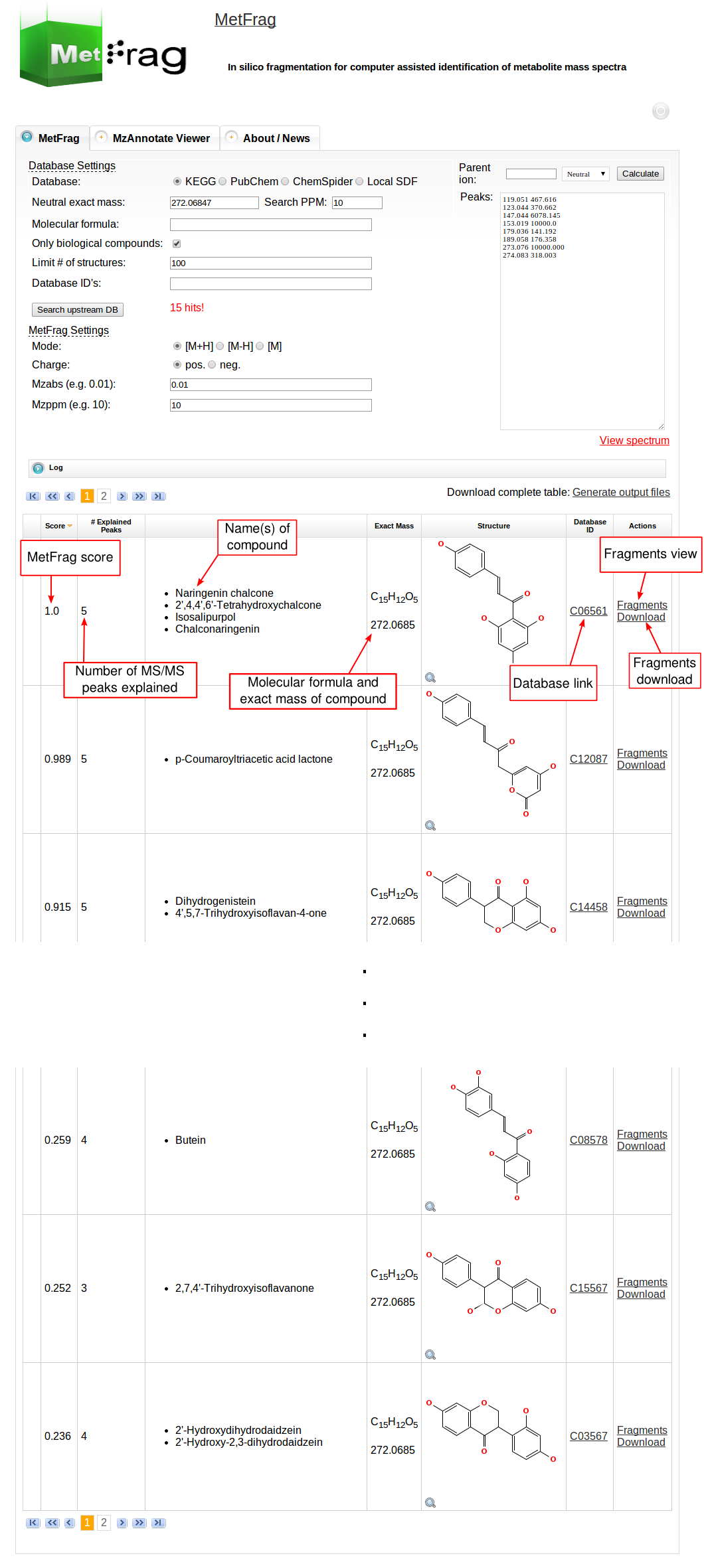
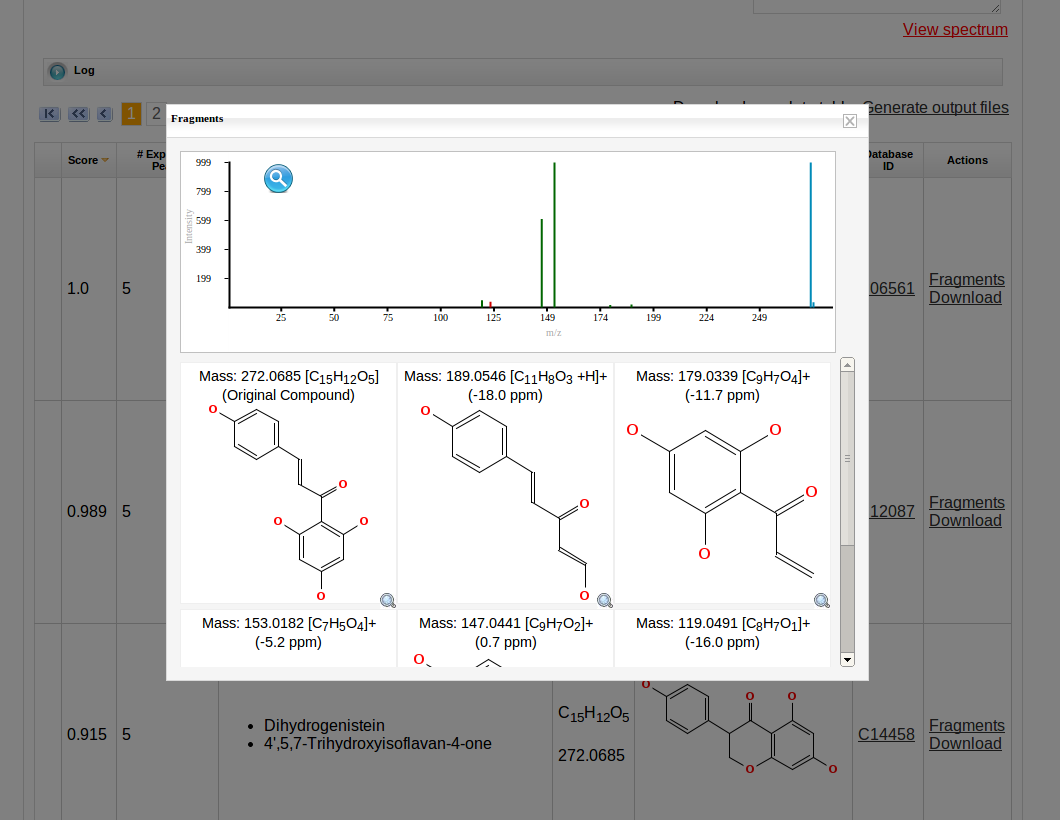
Then MetFrag starts the in silico fragmentation, the mapping of the fragments to the given mass peaks and the scoring for each retrieved candidate, respectively. Finally, the score ranked list of candidates will be displayed after the processing has finshed successfully. Each row in this list includes the score, number of explained peaks, name, molecular formula, monoisotopic mass, image and database identifier of the candiate. Additionally, in the last column links are given to download or display the fragments assigned to the given mass peaks for specific candidate. The database identifier is a external link to the entry of the selected database.