Discovering regulated Metabolite Families
Steffen Neumann
Leibniz Institute of Plant Biochemistrysneumann@ipb-halle.de
Guillaume Patoine
Leibniz Institute of Plant Biochemistrygpatoine@ipb-halle.de
Khabat Vahabi
Leibniz Institute of Plant Biochemistrydiscoveringregulatedmetabolitefamilies.RmdAbstract
Description of the MetFamily R package
MetFamily
MetFamily is an R package that provides a novel approach for the untargeted discovery of metabolite families.
Read more on the GitHub repository: https://github.com/ipb-halle/MetFamily
We first need to install the development version of the package.
remotes::install_github("ipb-halle/MetFamily@devel")Dataset
The following analysis looks into metabolites found in glandular trichomes of tomato plants. See the referenced paper for additional information [1].
The dataset is provided in the MetFamily package.
# MS1 intensities
filePeakMatrixPath <- system.file("extdata/showcase/Metabolite_profile_showcase.txt", package = "MetFamily")
# MS2 spectra
fileSpectra <- system.file("extdata/showcase/MSMS_library_showcase.msp", package = "MetFamily")
# Sirius annotations
fileAnnotation <- system.file("extdata/testdata/canopus/canopus1680.txt", package = "MetFamily")
# We use default import parameters
parameterSet <- parameterSetDefault()
parameterSet$minimumIntensityOfMaximalMS2peak <- 2000
parameterSet$minimumProportionOfMS2peaks <- 0.05
dataList <- projectFromFiles(
ms1_path = filePeakMatrixPath,
ms2_path = fileSpectra,
siriusFileColumnName = "ClassyFire class",
parameterSet = parameterSet,
annot_path = fileAnnotation
)## [1] "Parsing MS/MS file..."
## [1] "Parsing MS/MS file..."## MS/MS file: Read file## MS/MS file: Parse## MS/MS file: Filter## MS/MS file: Assemble spectra## [1] "Parsing MS/MS file ready"
## [1] "Parsing MS1 file..."
## [1] "Parsing MS1 file content..."
## [1] "Precursor deisotoping..."
## [1] "Precursor deisotoping 582 / 5823"
## [1] "Precursor deisotoping 1164 / 5823"
## [1] "Precursor deisotoping 1746 / 5823"
## [1] "Precursor deisotoping 2328 / 5823"
## [1] "Precursor deisotoping 2910 / 5823"
## [1] "Precursor deisotoping 3492 / 5823"
## [1] "Precursor deisotoping 4074 / 5823"
## [1] "Precursor deisotoping 4656 / 5823"
## [1] "Precursor deisotoping 5238 / 5823"
## [1] "Precursor deisotoping 5820 / 5823"
## [1] "Boxing..."
## [1] "Postprocessing matrix..."
## [1] "Building fragment mzFragmentGroups..."
## [1] "Fragment grouping preprocessing..."
## [1] "Fragment grouping preprocessing ready"
## [1] "Fragment grouping"
## [1] "Fragment grouping 34 / 86970"
## [1] "Fragment grouping 27083 / 86970"
##
## [1] "Fragment grouping ready (4.00752353668213s)"
## [1] "Fragment group postprocessing"
## [1] "Fragment group postprocessing: 1858 / 18589"
## [1] "Fragment group postprocessing: 3716 / 18589"
## [1] "Fragment group postprocessing: 5574 / 18589"
## [1] "Fragment group postprocessing: 7432 / 18589"
## [1] "Fragment group postprocessing: 9290 / 18589"
## [1] "Fragment group postprocessing: 11148 / 18589"
## [1] "Fragment group postprocessing: 13006 / 18589"
## [1] "Fragment group postprocessing: 14864 / 18589"
## [1] "Fragment group postprocessing: 16722 / 18589"
## [1] "Fragment group postprocessing: 18580 / 18589"
## [1] "Fragment group postprocessing ready (2.77388501167297s)"
## [1] "Boxing to matrix"
## [1] "Fragment group deisotoping"
## [1] "Fragment group deisotoping 1858 / 18589"
## [1] "Fragment group deisotoping 3716 / 18589"
## [1] "Fragment group deisotoping 5574 / 18589"
## [1] "Fragment group deisotoping 7432 / 18589"
## [1] "Fragment group deisotoping 9290 / 18589"
## [1] "Fragment group deisotoping 11148 / 18589"
## [1] "Fragment group deisotoping 13006 / 18589"
## [1] "Fragment group deisotoping 14864 / 18589"
## [1] "Fragment group deisotoping 16722 / 18589"
## [1] "Fragment group deisotoping 18580 / 18589"
## [1] "Fragment group deisotoping ready (13.2282903194427s)"
## [1] "Fragment group boxing"
## [1] "Building fragment mzFragmentGroups ready"## 'as(<dgCMatrix>, "dgTMatrix")' is deprecated.
## Use 'as(., "TsparseMatrix")' instead.
## See help("Deprecated") and help("Matrix-deprecated").## [1] "Boxing..."
## [1] "Ready"
## [1] "Preprocessing 473 / 2463"
## [1] "Preprocessing 1222 / 2463"
## [1] "Preprocessing 1965 / 2463"
## [1] "Features"
## [1] "Feature postprocessing"
## [1] "Coloring"
## [1] "Coloring init"
## [1] "Coloring naming functions"
## [1] "Coloring gather data"
## [1] "Coloring matrix"
## [1] "Feature annotations"
## [1] "Boxing"
## [1] "Ready"
## [1] "Merging by: featureId and Alignment ID"Filtering data
We use a filter for the intensity threshold and log2-fold-change to remove noise and focus on features of interest.
filterObj <- makeFilterObj(dataList, filter_average = 10000, filter_lfc = 2)PCA figures
pca <- calculatePCA(dataList=dataList,
filterObj=filterObj,
ms1AnalysisMethod="PCA (Principal Component Analysis)",
scaling="None",
logTransform=FALSE)## [1] "######################################################################################"
## [1] "PCA (Principal Component Analysis)"
## [1] "Analysis: pcaMethods"
resultObj <- calcPlotPCAscores(
pcaObj = pca,
dataList = dataList,
filterObj = filterObj,
showScoresLabels = FALSE,
xInterval = NULL,
yInterval = NULL
)
PCA scores of MS1
resultObj <- calcPlotPCAloadings(
pcaObj = pca,
dataList = dataList,
# filter = filterObj$filter, # deprecated
selectionFragmentPcaLoadingSet = NULL,
selectionAnalysisPcaLoadingSet = NULL,
selectionSearchPcaLoadingSet = NULL,
xInterval = NULL,
yInterval = NULL,
loadingsLabels = "None",
showLoadingsAbundance = FALSE,
showLoadingsFeaturesAnnotated = TRUE,
showLoadingsFeaturesUnannotated = TRUE,
showLoadingsFeaturesSelected = TRUE,
showLoadingsFeaturesUnselected = TRUE
)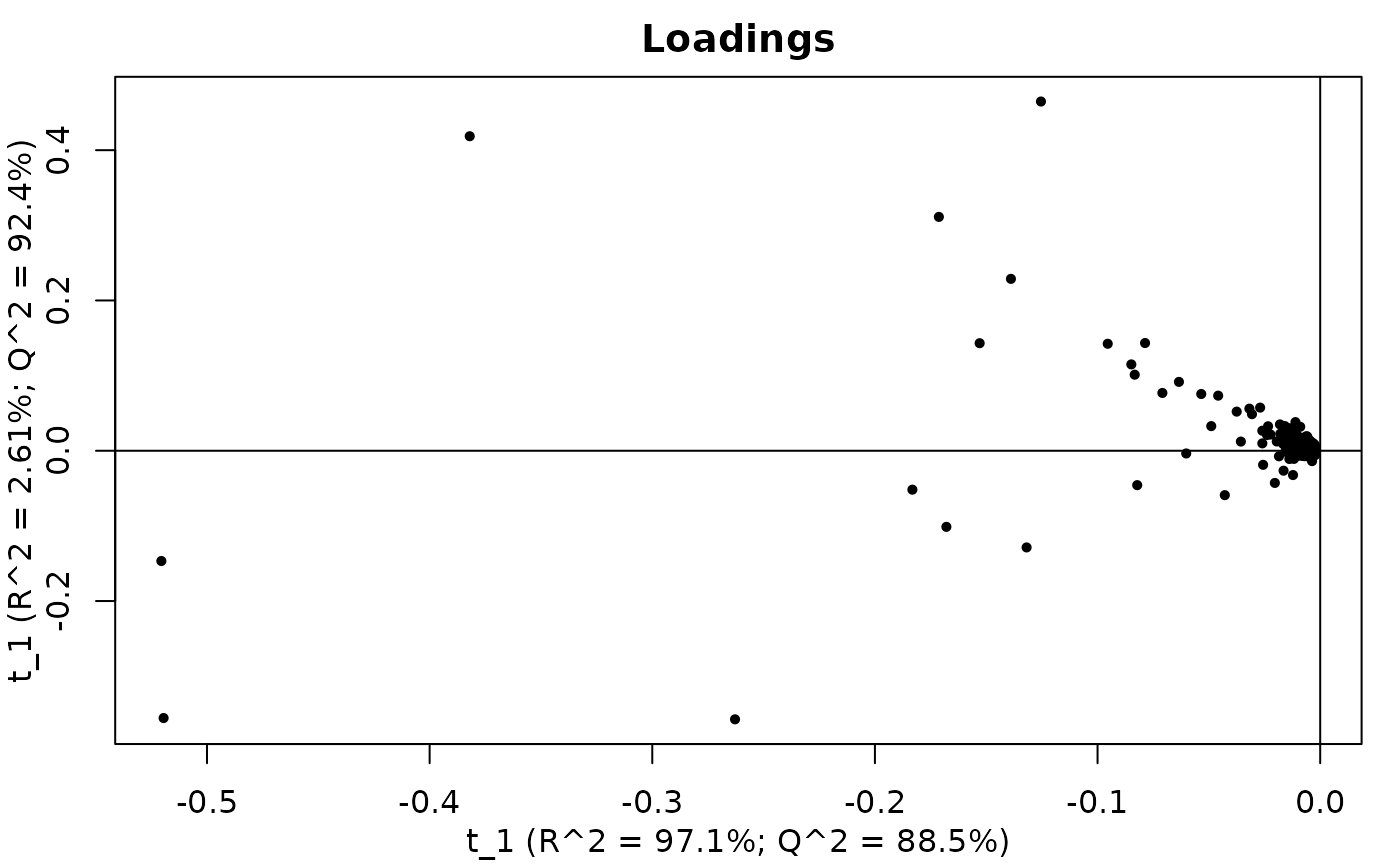
PCA loadings of MS1
HCA figure on MS2
In construction…
fileName <- system.file("extdata/testdata/clusterDataList.Rdata", package = "MetFamily")
load(fileName)
fileName <- system.file("extdata/testdata/hcaFilter.Rdata", package = "MetFamily")
load(fileName)
tryCatch(
returnObj <- calcPlotDendrogram(
dataList=dataList, #project,
filter=filter,
clusterDataList=clusterDataList,
annoPresentAnnotationsList = annoPresentAnnotationsList ,
annoPresentColorsList = annoPresentColorsList,
distanceMeasure="Jaccard (intensity-weighted)",
selectionFragmentTreeNodeSet = NULL,
selectionAnalysisTreeNodeSet = NULL,
selectionSearchTreeNodeSet = NULL,
showClusterLabels = TRUE,
hcaPrecursorLabels = "m/z / RT",
xInterval = c(1,219)),
error = \(x){invisible(NULL)}
)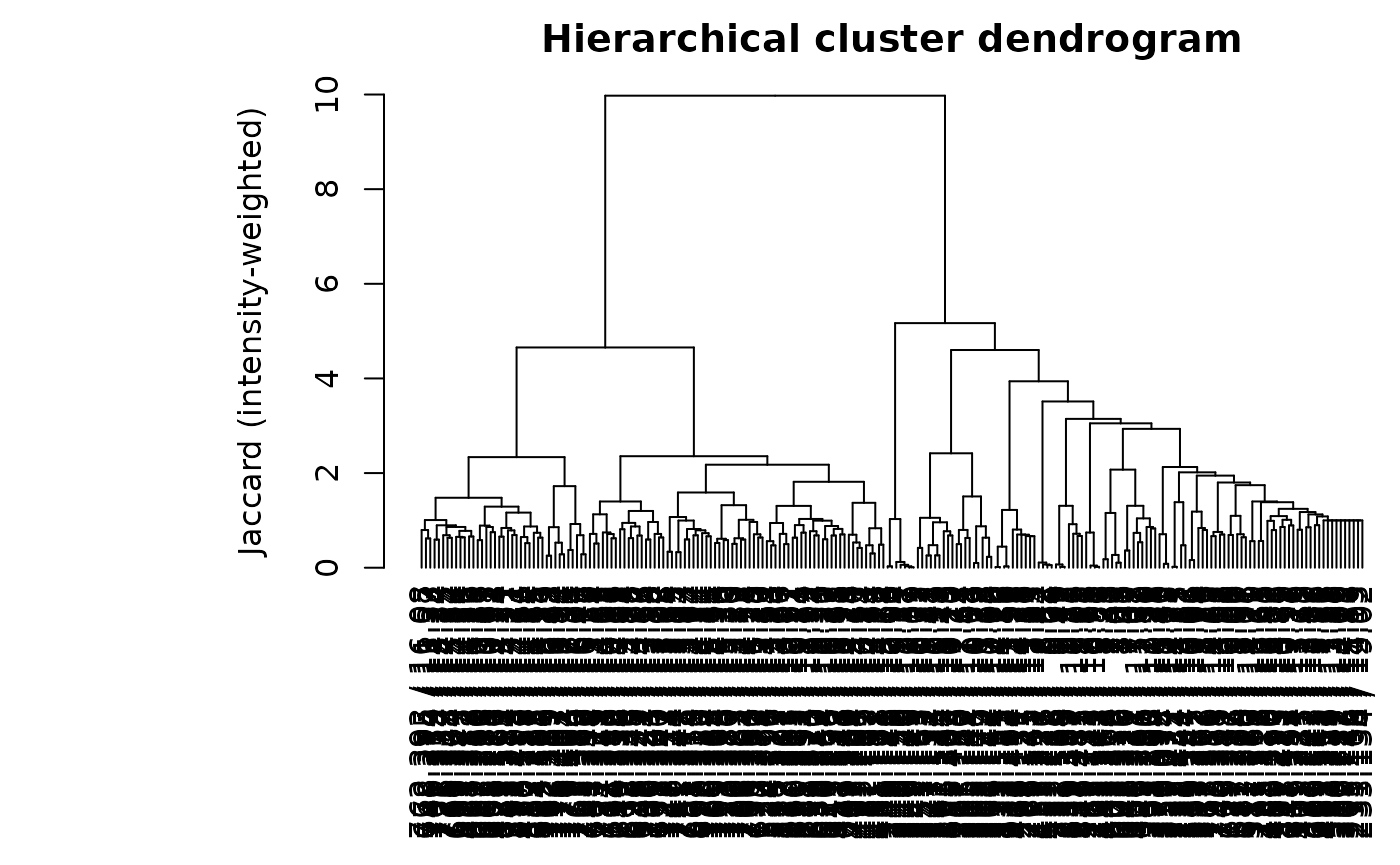
HCA on MS2
References
1. Treutler H, Tsugawa H, Porzel A, Gorzolka K, Tissier A, Neumann S,
Balcke GU: Discovering
Regulated Metabolite Families in
Untargeted Metabolomics
Studies. Anal Chem 2016.